2022_New Research Page for Website
The Smogorzewska laboratory seeks to understand how DNA repair that occurs during DNA replication contributes to normal organ function. The main paradigm we use is the genetic diseases that are associated with the deficiency of DNA repair during replication with the primary effort concentrated on understanding the DNA interstrand crosslink (ICL) repair.
ICL repair takes place at sites where the two strands of the DNA have become covalently linked by the byproducts of lipid peroxidation, endogenous aldehydes, and as-yet unknown metabolites. The repair of these lethal lesions requires a dual excision of the crosslinked bases and repair of the resulting double-strand breaks.


This feat is accomplished in a multi-step process mediated by the Fanconi anemia (FANC) proteins including familial breast and ovarian proteins BRCA1 and BRCA2.
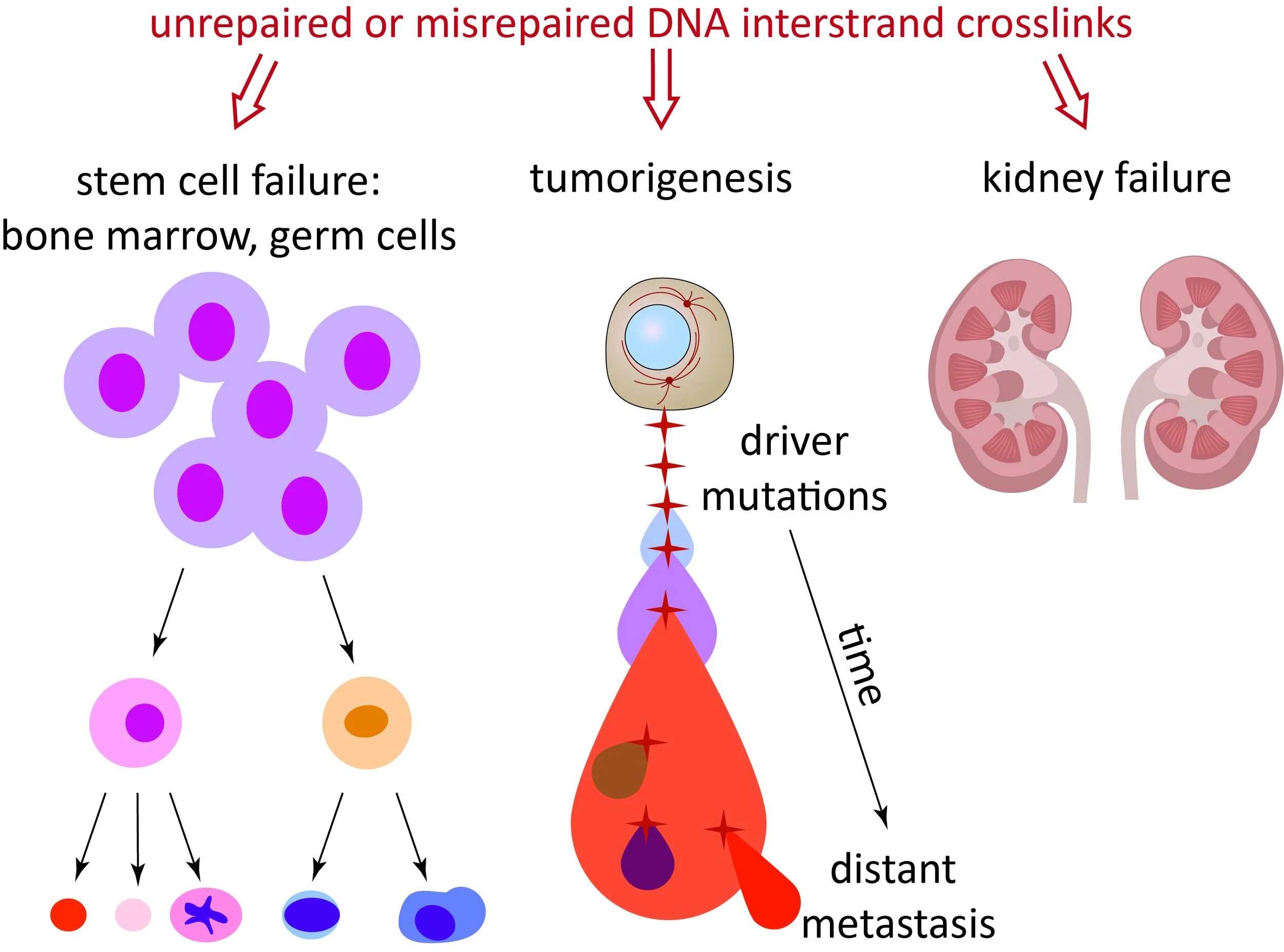
Fanconi anemia (FA) patients lack components of this pathway, through mutations in the FANC genes, and suffer from developmental abnormalities, bone marrow failure, and infertility due to the inability to maintain stem cell function. FA is also associated with a very high incidence of cancer, due to the mutagenic nature of misrepaired ICLs. On the other hand, the induction of ICLs is a major method of cancer treatment in sporadic cancers and often leads to excellent outcomes (e.g. cisplatin treatment of testicular cancer). ICL repair is of critical importance to normal development, tissue homeostasis, and in the context of cancer.
While we continue to study the functions of specific proteins in the pathway, which is essential to understand the molecular mechanism of ICL DNA repair, over the last few years we have positioned ourselves to study the pathogenesis of the genome instability disorders with the goal of identifying therapeutic approaches and targets in these diseases.
Our current work is motivated by three themes:
UNDERSTANDING TUMORIGENESIS IN FANCONI AMEMIA
IDENTIFYING ENDOGENOUS SOURCES OF DNA DAMAGE
UNDERSTANDING DNA REPLICATION AND REPAIR
Understanding tumorigenesis in Fanconi anemia
The first theme is to understand the process of tumorigenesis in Fanconi anemia, both the aerodigestive tract squamous cell carcinomas and the embryonal tumors including medulloblastoma.
The goal is to understand the basic biology of these largely unstudied FA cancers to improve diagnosis and treatment, to identify synthetic interactions with the genetic vulnerabilities present in these cancers, to develop patient derived and animal models of the diseases and eventually to test therapeutic approaches.
Squamous cell carcinoma tumorigenesis in Fanconi anemia

Examples of patient samples with the top left picture showing normal mucosa, the middle top picture showing transition of normal to cancerous area, and the other pictures showing tumors.

By sequencing squamous cell carcinomas from Fanconi anemia patients, we demonstrate that the primary genomic signature of Fanconi anemia deficiency is the presence of a high number of structural variants. Structural variants are enriched for small deletions, unbalanced translocations, and fold-back inversions that arise in the context of TP53 loss.
Resultant genomic instability underlies elevated somatic copy number alteration (sCNA) rates of key HNSCC-associated genes, including PIK3CA, MYC, CSMD1, PTPRD, YAP1, MXD4, and EGFR.
In contrast to sporadic HNSCC, we find no evidence of HPV infection in FA HNSCC, although positive cases were identified in gynecologic tumors.
Using proteogenomic and digital histologic analysis to compare sporadic HNSCC and FA-related HNSCC we can identify common and cohort-specific therapies.
FA-deficient tumors concurrently undergo swift epithelial to mesenchymal transition (EMT) as indicated by changes in cell morphology, the upregulation of mesenchymal genes, and the downregulation of epithelial genes. Furthermore, transcriptomic analysis of the FA-deficient tumors shows robust activation of an inflammatory response. We aim to understand the mechanism of the epithelial to mesenchymal transition priming in FA and immune activation in the setting of DNA damage.



BRCA2 is an essential DNA repair protein whose heterozygous deficiency leads to familial breast, ovarian, prostate, and pancreatic cancers. 70% of patients with biallelic mutations in BRCA2 develop medulloblastoma, the most common childhood brain malignancy, before the age of five and succumb to the disease because of lack of treatment options. The mechanism of how BRCA2 prevents tumor formation is not clear especially in neurons of the cerebellum. We hypothesize that there exist endogenous sources of DNA lesions that necessitate function of BRCA2 and that the lack of BRCA2 in an already formed tumor may make them susceptible to loss of other DNA repair pathways. By tumor sequencing and functional analysis of tumor-derived cells, we aim to understand the pathogenesis of tumors without BRCA2 function.
Identifying endogenous sources of DNA damage

The second theme is to identify the endogenous sources of DNA damage that determine why specific tissues/stem cells fail in the disorders of ICL repair deficiency. The hypothesis is that different tissues have different sources of endogenous DNA damage that require ICL repair proteins.
Understanding what they are and how they are produced will give us an opportunity to manipulate them to prevent/delay the onset of the disease manifestations.
Using CRISPR screens, we aim to identify endogenous sources of DNA damage in different tissues, including hematopoietic stem cells and keratinocytes.
Publication: Blood, Jung 2021 (review)
Understanding DNA replication and repair
The third theme is to understand the fundamental events at the crossroads of DNA replication and repair. Our studies of Fanconi anemia and related diseases brought to our attention the vast gaps of knowledge about the mechanisms of cellular response to challenges during DNA replication. With human replisomes moving just under 1000 nucleotides per minute and replicating DNA with great fidelity, the response of the replisome to any kind of challenge needs to be very dynamic and superbly regulated.

Our laboratory has previously shown that proteasome shuttle proteins, DNA Damage Inducible 1 (DDI1) and DNA Damage Inducible 2 (DDI2), function to remove Replication Termination Factor 2 (RTF2) from the stalled replisome allowing for replication fork stabilization and restart during replication stress response. Since the publication of this study, we have explored the normal function of RTF2 and have been making strides in identifying the physiological reason for its removal from the replisome.
We were able to show that lack of RNASE H2 also led to slow replication speeds presumably due to the increased levels of ribonucleotides that are embedded in the DNA and we are currently teasing out the regulation of RTF2-RNASE H2 interaction, both genetically and biochemically.
Another goal of this work is to leverage the idea that the removal of proteins during DNA damage response (DDR) is as equally important as recruitment of DNA repair proteins to sites of DNA damage.
Schematic showing our model of RTF2 function during normal replication and in response to replication stress. We determined that during active replication, RTF2 travels with the replisome and is necessary for normal replication speed partially through regulation of RNAse H2, enzyme that removes DNA-embedded nucleotides. Upon replication stress, RTF2 is removed allowing replication restart using priming activity.
Previous Work
Our previous published work has brought new findings including identification of genes necessary for interstrand crosslink (ICL) repair: FANCP/SLX4, FANCR/RAD51, and FANCT/UBE2T.
We have also identified FAN1 in an shRNA screen for proteins necessary for ICL repair and showed that FAN1 deficiency was responsible for the cellular defects in patients with Karyomegalic Interstitial Nephritis (KIN).
IDENTIFICATION OF GENES MUTATED IN ICL REPAIR DISORDERS




Patient cell lines are invaluable to identify mechanisms of ICL repair. Our lab is uniquely positioned to apply advanced sequencing technologies on patient samples from the International Fanconi Anemia Registry (IFAR) at Rockefeller University. IFAR was established in 1982 by Dr. Arleen Auerbach.
Especially useful are identifying naturally occurring separation of function mutations that direct us to new functions of proteins.


WHAT IS THE PHYSIOLOGICAL ROLE OF FAN1?

We have made a mouse model of FAN1 deficiency and are studying the phenotypes of this mouse to gain better understanding of human disease and the in vivo consequences of inappropriate ICL repair.

